Виртуелен кариотип
Виртуелен кариотип ― дигитална информација што одразува кариотип, што произлегува од анализата на кратки секвенци на ДНК од специфични места низ целиот геном, кои се изолирани и набројани.[1] Открива варијации на бројот на геномски копии со повисока резолуција за ниво од конвенционалната кариотипизација или споредбената геномска хибридизација засновата на хромозом.[2] Главните методи што се користени за создавање виртуелни кариотипови се низа на споредбената геномска хибридизација и низа на единечно нуклетидниот полиморфизам.
Заднина
уредиКариотипот (слика 1) е карактеристичниот хромозомски комплемент на еден еукариотски вид.[3][4] Кариотипот обично се прикажуван како слика на хромозомите од една единствена клетка распоредена од најголем (хромозом 1) до најмал (хромозом 22), а последни се прикажани половите хромозоми (X и Y). Историски гледано, кариотиповите се добивани со боење на клетките откако тие биле хемиски прекинати за време на клеточната делба. Кариотиповите се користени неколку децении за да бидат идентификувани хромозомските абнормалности и во микробните и во ракородните клетки. Конвенционалните кариотипови можат да го проценат целиот геном за промени во структурата и бројот на хромозомите, но резолуцијата е релативно груба, со ограничување за откривање од 5-10 Mb.[се бара извор]

Метод
уредиНеодамна, биле појавени платформи за создавање кариотипови, правени на сметач и со висока резолуција од нарушена ДНК, како што се низа на споредбена геномска хибридизација и низа на единечна нуклеотидна полиморфизам. Концептуално, низите се составени од стотици до милиони сонди кои се комплементарни на регионот од интерес во геномот. Нарушената ДНК од тест примерокот е расцепкана, означена и хибридизирана во низата. Интензитетот на сигналот за хибридизација за секоја сонда е користен од специјализиран софтвер за да биде направен log2 соодносен од примерок и контрола за секоја сонда на низата.[се бара извор]
Знаејќи ја местоположбата на секоја сонда на низата и местоположбата на секоја сонда во геномот, софтверот ги подредува сондите во хромозомски редослед и го реконструира геномот дигитално (слика 2).
Виртуелните кариотипови имаат драматично повисока резолуција од конвенционалната цитогенетика. Вистинската резолуција ќе зависи од густината на сондите на низата. Во моментов, Affymetrix SNP6.0 е комерцијално достапна низа со најголема густина за применувања за виртуелна кариотипизација. Содржи 1,8 милиони полиморфни и неполиморфни маркери за практична резолуција од 10–20 kb - приближно со големина на ген. Ова е приближно 1000 пати поголема резолуција од кариотиповите добиени од конвенционалната цитогенетика.[се бара извор]
Виртуелните кариотипови може да бидат изведени на примероци од микробната линија за конституционални нарушувања,[5][6] и клиничкото тестирање е достапно од десетици лаборатории сертифицирани со CLIA (genetests.org). Виртуелна кариотипизација може да биде направена и на свежи или формалинско фиксирани парафински тумори.[7][8][9] Лабораториите кои се сертифицирани со CLIA, кои нудат тестирање на тумори се Creighton Medical Laboratories Архивирано на 11 февруари 2010 г. (свежи и парафински вградени примероци на тумор) и CombiMatrix Molecular Diagnostics (свежи примероци на тумор).

Различни платформи за виртуелно кариотипирање
уредиКариотипизацијата заснована на низа може да се направи со неколку различни платформи, и лабораториски развиени и комерцијални. Самите низи можат да бидат ширум геномот (сонди распространети низ целиот геном) или насочени (сонди за геномски региони за кои е познато дека се вклучени во одредена болест) или комбинација од двете. Понатаму, низите што се користени за кариотипизација може да користат неполиморфни сонди, полиморфни сонди (т.е. што содржат единечен нуклеотиден полиформизам) или комбинација од двете. Неполиморфните сонди можат да обезбедат само информации за бројот на копијата, додека низите на единечен нуклеотиден полиформизам можат да обезбедат и број на копии и статус на загуба на хетерозиготност во една анализа. Врстите на сонди кои се користени за неполиморфни низи се копирана, клонови на бактериски вештачки хромозоми (на пр. BlueGnome Архивирано на 7 февруари 2014 г.) и олигонуклеотиди (на пример, Agilent, Санта Клара, Калифорнија, Соединетите Држави или Nimblegen, Медисон, Висконсин, Соединетите Држави). Комерцијално достапните олигонуклеотидни низи на единечниот нуклеотиден полиформизам можат да бидат со цврста фаза (Affymetrix, Санта Клара, Калифорнија) или врз основа на монистра (Illumina, Сан Диего, Калифорнија). И покрај разновидноста на платформите, на крајот сите тие користат геномска ДНК од нарушените клетки за да создадат дигитален кариотип со висока резолуција. Крајниот производ сè уште нема доследно име и е наречен виртуелно кариотипирање,[8][10] дигитално кариотипирање,[11] молекуларна алелокариотипизација,[12] и молекуларна кариотипизација.[13] Други поими што се користени за опишување на низите што се користат за кариотипизација вклучуваат олигонуклеотидни микрочипови со единечен нуклеотиден полиформизам[14] и хромозомски микрочип.[15][16] Некои сметаат дека сите платформи се вид на низи на споредбена геномска хибридизација, додека други го задржуваат тој поим за методите со две бои, а трети ги издвојуваат низите на единечен нуклеотиден полиформизам бидејќи тие создаваат повеќе и различни информации од методите со двобојна низа на споредбената гемонска хибридизација.
Примени
уредиОткривање на промени во бројот на копии
уредиПромените на бројот на копии може да се видат и во примероците од микробните и туморските примероци. Промените на бројот на копии може да бидат забележани со низи со неполиморфни сонди, како што е низа на споредбената геномска хибридизација, и со низи засновани на единечниот нуклеотиден полиформизам. Луѓето се диплоидни, така што нормалниот број на копии е секогаш два за неполовите хромозоми.[се бара извор]
- Бришење: Бришење е губење на генетски материјал. Бришењето може да биде хетерозиготно (копија број 1) или хомозиготно (копија број 0, нулисомија). Микроделеционите синдроми се примери на конституционални нарушувања поради мали бришења во ДНК на микробната линија. Бришењето во клетките на туморот може да претставува исклучување на туморски потиснувачки ген и може да има дијагностички, прогностички или терапевтски последици.
- Добивки: добивката на бројот на копијата ја претставува добивката на генетскиот материјал. Ако добивката е од само една дополнителна копија на сегмент од ДНК, тоа може да биде наречено удвојување (сл. 3). Ако има една дополнителна копија од цел хромозом, тоа може да биде наречено трисомија. Зголемувањето на бројот на копии во примероците од микробната линија може да биде поврзано со болеста или може да биде бенигна варијанта на бројот на копии. Кога се гледани во клетките на туморот, тие може да имаат дијагностички, прогностички или терапевтски последици.

- Засилувања: Технички, засилувањето е тип на зголемување на бројот на копијата во која има број на копирање >10. Во контекст на биологијата на ракот, често се забележувани засилувања кај онкогените. Ова може да укаже на полоша прогноза, да помогне да се категоризира туморот или да укаже на подобност за лекови. Пример за подобност на лекот е засилувањето Her2Neu и Herceptin, а дадена е слика на засилување Her2Neu откриена со виртуелно кариотипирање на низата на единечен нуклеотиден полиформизам (сл. 4).
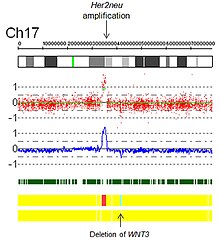
Слика 4. Засилувањео со Her2 со низа на единечен нуклеотиден полиформизам од виртуелен кариотип.

Губење на хетерозиготност, автозиготни сегменти и еднородителска дисомија
уредиАвтозиготните сегменти и еднородителната дисомија се диплоидни/„кописко неутрални“ генетски наоди и затоа се забележувани само со низи засновани на единечен нуклеотиден полиформизам. И автозиготните сегменти и еднородителската дисомија ќе покажат губење на хетерозиготност со број на копии од два со кариотипирање на низата на единечниот нуклеотиден полиформизам. Поимот истеци на хомозгиготност (Runs of Homozgygosity) е општи поим кој може да биде користен или за автозиготни сегменти или за еднородителска дисомија.
- Автозиготен сегмент: Автозиготниот сегмент е двородител и е гледан само во микробната линија. Тие се продолжени низи на хомозиготни маркери во геномот и се појавуваат кога идентичен хаплотипски блок е наследен од двата родители. Тие се нарекувани и сегменти „идентични по потекло“ и можат да бидат користени за картирање на хомозиготноста.[17][18]
- Еднородителска дисомија: Еднородителската дисомија се јавува кога двете копии на ген или геномски регион се наследени од истиот родител. Ова е еднородителско, за разлика од автозиготните сегменти кои се двородителски. Кога се присутни во микробната линија, тие можат да бидат безопасни или поврзани со болести, како што се Прадер-Вилиевиот или Ангелмановиот синдром. Исто така, за разлика од автозиготноста, еднородителската дисомија може да биде развиена во клетките на туморот, и тоа е нарекувано во книжевноста како стекната еднородителска дисомија или копирање неутрална загуба на хетерозиготност. Стекнатата еднородителска дисомија е доста честа појава и кај хематолошките и кај цврстите тумори и е пријавено дека сочинува 20 до 80% од загубата на хетерозиготност забележан кај човечките тумори.[19][20][21][22] Стекнатата еднородителска дисомија може да послужи како втор погодок во Кнадсоновата хипотеза за туморска генеза со два погодоци, и на тој начин може да биде биолошки еквивалент на бришење.[23] Бидејќи оваа врста на лезија не може да се открие со низата на споредбената геномска хибридизација, флуоресцентната „на лице место“ хибридизација или конвенционална цитогенетика, се претпочитаат низи засновани на единечен нуклеотиден полиформизам за виртуелна кариотипизација на туморите.
Примери на клинички примени против рак
уредиВиртуелен кариотип може да биде создаден од речиси секој тумор, но клиничкото значење на идентификуваните геномски аберации се различни за секој вид на тумор. Клиничката корист варира и соодветноста најдобро ја одредува онколог или патолог во советување со лабораторискиот директор на лабораторијата што го изведува виртуелниот кариотип. Подолу се дадени примери на типови на рак каде клиничките последици на специфичните геномски аберации се добро утврдени. Овој список е претставителен, не е исцрпувачки. Мрежната страница на Лабораторијата за цитогенетика во Државната лабораторија за хигиена во Висконсин има дополнителни примери на клинички релевантни генетски промени кои лесно се откривамо со виртуелна кариотипизација.[1] Архивирано на 12 април 2009 г.
Невробластом
уредиВрз основа на серија од 493 примероци на невробластом, објавено е дека севкупниот геномски модел, како што е тестиран со кариотипизација заснован на низа, е претскажувач за исходот кај невробластомот:[24]
- Туморите кои се манифестирале исклучиво со промени на бројот на копии на цела хромозом биле поврзани со одлично преживување.
- Туморите кои се манифестираат со било каков вид на промени на бројот на копијата на сегменталниот хромозом биле поврзани со висок ризик од релапс.
- Во рамките на туморите кои покажуваат сегментални промени, дополнителни независни предиктори за намаленото вкупно преживување биле засилување со генот MYCN, бришења на 1p и 11q, и добивање на 1q.
Претходните публикации ги категоризирале невробластомите во три главни подвидови врз основа на цитогенетските профили:[25]
- Подвид 1: поволен невробластом со речиси триплоидија и доминација на бројчани добивки и загуби, кои претежно претставуваат неметастатски невробластомски фази 1, 2 и 4S.
- Подвидови 2A и 2B: пронајдени во неповолен широко распространет невробластом, фази 3 и 4, со загуба на 11q и засилување на 17q засилување без засилување на генот MYCN (подвид 2A) или со засилување на MYCN често заедно со бришење на 1p и засилување на 17q (подвид 2B).
Вилмсов тумор
уредиГубењето на хетерозиготност кое се однесува само на тумори, за хромозомите 1p и 16q идентификува подгрупа на пациенти со Вилмсовиот тумор кои имаат значително зголемен ризик од релапс и смрт. Загубата на хетерозиготност за овие хромозомски региони сега може да биде користен како независен прогностички фактор заедно со фазата на болеста за да се насочи интензитетот на третманот до ризикот од неуспех на третманот.[26][27]
Бубрежно-клеточен карцином
уредиБубрежните епителни неоплазми имаат карактеристични цитогенетски аберации кои можат да помогнат во класификацијата.[28] Поврзано Атлас за генетика и цитогенетика во онкологијата и хематологијата.
- Јасно-клеточен карцином: губење на 3p
- Папиларен карцином: трисомија 7 и 17
- Хромофобен карцином: хиподиплоиден со губење на хромозомите 1, 2, 6, 10, 13, 17, 21
Кариотипизацијата заснована на низа може да биде користена за да бидат идентификувани карактеристичните хромозомски аберации кај бубрежните тумори со предизвикувачка морфологија.[8][10] Кариотипизацијата заснована на низа работи добро на тумори вградени во парафин[29] и е подложна на рутинска клиничка употреба.
Дополнително, неодамнешната книжевност покажува дека одредени хромозомски аберации се поврзани со исходот кај специфични подтипови на бубрежни епителни тумори.[30] Јасно-клеточен бубрежен карцином: del 9p и del 14q се лоши прогностички показатели.[31][32] Папиларен карцином на бубрежни клетки: удвојување на 1q означува фатален напредок.[33]
Хронична лимфоцитна леукемија
уреди
Кариотипизацијата заснована на низа е исплатлива алтернатива на флуоресцентната „на лице место“ хибридизација за откривање на хромозомски абнормалности кај хронична лимфоцитна леукемија. Неколку клинички студии за валидација покажале >95% усогласеност со стандардниот панел на хроничната лимфоцитна леукемија со флуоресцентната „на лице место“ хибридизација.[12][34][35][36][37] Дополнително, многу студии кои користат кариотипизација заснована на низа идентификуваа „невообичаени бришења“ пропуштени од стандардните сонди со флуоресцентната „на лице место“ хибридизација и стекнати унипарентална дисомија на клучните локуси за прогностички ризик кај хроничната лимфоцитна леукемија.[38][39]
Во клетките на хроничната лимфоцитна леукемија се препознаени четири главни генетски аберации кои имаат големо влијание врз однесувањето на болеста.[40]
- Бришењето на дел од краткиот крак на хромозомот 17 (del 17p) кои се насочени кон p53 се особено штетни. Пациентите со оваа абнормалност имаат значително краток интервал пред да им биде потребна терапија и пократко преживување. Оваа абнормалност се наоѓа кај 5-10% од пациентите со хроничната лимфоцитна леукемија.
- Бришењето на долгата рака на хромозомот 11 (del 11q) се исто така неповолни, иако не до степенот забележан со дел 17p. Абнормалноста го таргетира генот ATM и се јавува ретко кај CLL (5-10%).
- Трисомија 12, дополнителен хромозом 12, е релативно чест наод што се јавува кај 20-25% од пациентите и дава средна прогноза.
- Бришењето на 13q14 (del 13q14) е најчеста абнормалност кај хроничната лимфоцитна леукемија кај приближно 50% од пациентите со клетки кои го содржат овој дефект. Кога del 13q14 се гледа изолирано, пациентите имаат најдобра прогноза и повеќето ќе живеат многу години, дури и децении, без потреба од терапија.
Повеќекратна миелома
уредиАвет-Лоазо и колегите, во Journal of Clinical Oncology, користеле кариотипизација на единечниот нуклеотиден полиморфизам низа од 192 примероци на повеќекратна миелома за да бидат идентификувани генетските лезии поврзани со прогнозата, кои потоа биле потврдени во посебна група (n = 273).[41] Кај повеќекратната миелома, недостатокот на пролиферативен клон ја прави конвенционалната цитогенетика информативна само во ~ 30% од случаите. Панелите со флуоресцентната „на лице место“ хибридизација се корисни во повеќекратната миелома, но стандардните панели нема да забележуваат неколку клучни генетски абнормалности пријавени во оваа студија.
- Виртуелната кариотипизација идентификувала хромозомски абнормалности во 98% од случаите на повеќе кратната миелома
- del(12p13.31) е независен несакан маркер
- amp (5q31.1) е поволен маркер
- Прогностичкото влијание на amp (5q31.1) го надминува она на хипердиплоидијата и исто така ги идентификува пациентите кои имаат голема корист од терапијата со високи дози.
Кариотипирањето засновано на низа не може да открие урамнотежени транслокации, како што е t(4;14) забележано кај ~15% од повеќекратната миелома. Затоа, флуоресцентната „на лице место“ хибридизација за оваа транслокација, исто така, треба да се изведе ако се користени низи со единечен нуклеотиден полиморфизам за откривање на промени на бројот на копии ширум геномот од прогностичко значење кај повеќекратната миелома.
Медулобластом
уредиКариотипизација на 260 медулобластоми врз основа на низа од Штефан Пфистер и колегите, резултирало со следните клинички подгрупи врз основа на цитогенетските профили:[42]
- Лоша прогноза: зголемување на 6q или засилување на MYC или MYCN
- Средно: засилување од 17q или i(17q) без засилување од 6q или засилување на MYC или MYCN
- Одлична прогноза: урамнотежени 6q и 17q или бришење на 6q
Олигодендроглиом
уредиЗаемното бришење 1p/19q е сметано за „генетски потпис“ на олигодендроглиом. Алелните загуби на 1p и 19q, одделно или комбинирани, се почести кај класичните олигодендроглиоми отколку кај астроцитомите или олигоастроцитомите.[43] Во една студија, класичните олигодендроглиоми покажале 1p загуба во 35 од 42 (83%) случаи, 19q загуба во 28 од 39 (72%), и тие биле комбинирани во 27 од 39 (69%) случаи; немало значајна разлика во 1p/19q губење на статусот на хетерозиготност помеѓу низок степен и анапластични олигодендроглиоми.[43] Заемното бришење на 1p/19q е во корелација и со хемосензитивност и со подобрена прогноза кај олигодендроглиомите.[44][45] Повеќето поголеми центри за третман на рак рутински проверуваат за бришење на 1p/19q како дел од извештајот за патологија за олигодендроглиоми. Статусот на локусите 1p/19q може да биде откриен со флуоресцентната „на лице место“ хибридизација или виртуелно кариотипирање. Виртуелното кариотипирање ја има предноста да го процени целиот геном во една анализа, како и локусите 1p/19q. Ова овозможува проценка на другите клучни локуси во глијалните тумори, како што се статусот на бројот на копијата на EGFR и TP53.
Додека прогностичката релевантност на бришењата 1p и 19q е добро воспоставена за анапластични олигодендроглиоми и мешани олигоастроцитоми, прогностичката релевантност на бришењата за глиоми со низок степен е поконтроверзна. Во однос на глиоми со низок степен, една неодамнешна студија, исто така, сугерира дека заемното бришење на 1p/19q може да биде поврзано со (1;19)(q10;p10) транслокација која, како и комбинираното бришење на 1p/19q, е поврзана со супериорна целокупно преживување и преживување без напредок кај пациенти со низок степен на глиома.[46] Олигодендроглиомите покажуваат само ретко мутации во генот p53, што е во спротивност со другите глиоми.[47] Засилувањето на рецепторот на епидермалниот фактор на раст и целото заемно бришење на 1p/19q меѓусебно се исклучуваат и предвидуваат сосема различни исходи, при што засилувањето на EGFR предвидува лоша прогноза.[48]
Глиобластом
уредиЈин и колегите[49] проучувал 55 глиобластоми и 6 клеточни линии на глиобластомски многуоблици користејќи кариотипизација на низа на единечен нуклеотиден полиморфизам. Стекнатата еднородителска дисомија била идентификувана на 17p во 13/61 случаи. Било пронајдено значително скратено време на преживување кај пациенти со бришење 13q14 (RB) или бришење/стекната еднородителска дисомија со 17p13.1 (p53). Земени заедно, овие резултати наведуваат дека оваа техника е брз, робустен и евтин метод за профилирање на абнормалности на геномот во глиобластомските многуоблици. Бидејќи кариотипизацијата на низата на единечен нуклеотиден полиморфизам може да биден извршена на тумори вградени во парафин, тоа е привлечен можност кога клетките на туморот не успеваат да растат во културата за метафазна цитогенетика или кога желбата за кариотипизација се јавува откако примерокот е фиксиран со формалин.
Важноста на откривање на стекнатата еднородителска дисомија (копирање на неутрална загуба на хетерозиготност) кај глиобластом:
- Од пациентите со 17p абнормалности, ~50% биле бришења и ~50% биле стекната еднородителска дисомија
- И еднородителската дисомија на 17p del и 17p биле поврзани со полош исход.
- 9/13 имаше хомозиготни TP53 мутации во основата на еднородителската дисомија на 17p.
Дополнително, во случаи со несигурна оценка по морфологија, геномското профилирање може да помогне во дијагнозата.
- Истовремената добивка од 7 и загуба од 10 е суштински патогномонична за глиобластомски повеќеоблици[50]
- Засилување на EGFR, губење на PTEN (на 10q) и губење на p16 (на 9p) се јавуваат речиси исклучиво кај глиобластом и може да обезбеди средства за разликување на анапластичниот астроцитом од глиобластом.[51]
Акутна лимфобластична леукемија
уредиЦитогенетиката, проучувањето на карактеристичните големи промени во хромозомите на клетките на ракот, се повеќе е препознавана како важен претскажувач за исходот кај акутната лимфобластична леукемија.[52] Невробластом: Урамнотежените транслокации не можат да бидат откриени со кариотипирање засновани на низа (види Ограничувања подолу).
Некои цитогенетски подвидови имаат полоша прогноза од другите. Тие се:
- Транслокација помеѓу хромозомите 9 и 22, позната како филаделфискиот хромозом, се јавува кај околу 20% од возрасните и 5% кај педијатриските случаи на акутната лимфобластична леукемија.
- Транслокација помеѓу хромозомите 4 и 11 се јавува во околу 4% од случаите и е најчеста кај доенчиња под 12 месеци.
- Не сите транслокации на хромозоми носат полоша прогноза. Некои транслокации се релативно поволни. На пример, хипердиплоидијата (> 50 хромозоми) е добар прогностички фактор.
- Процената на промените на бројот на копии во геном може да се направи со конвенционална цитогенетика или виртуелна кариотипизација. Виртуелното кариотипирање на низата на единечниот нуклеотиден полиморфизам може да открие промени на бројот на копии и статусот на загуба на хетерозиготност, додека низата на споредбената геномска хибридизација може да открие само промени на бројот на копии. Копирање на неутрална загуба на хетерозиготност (стекната еднородителска дисомија) е пријавена во клучните локуси во акутната лимфобластична леукемија, како што е генот CDKN2A на 9p, кои имаат прогностичко значење.[53][54][55] Виртуелното кариотипирање на низата на единечниот нуклеотиден полиморфизам може лесно да открие копирање на неутрална загуба на хетерозиготност. Низата на споредбена геномска хибридизација, флуоресцентната „на лице место“ хибридизација и конвенционалната цитогенетика не можат да забележуваат копирање на неутрална загуба на хетерозиготност.
| Цитогенетска промена | Категорија на ризик |
|---|---|
| Филаделфиски хромозом | Лоша прогноза |
| t(4;11)(q21;q23) | Лоша прогноза |
| t(8;14)(q24.1;q32) | Лоша прогноза |
| Комплексен кариотип (повеќе од четири абнормалности) | Лоша прогноза |
| Ниска хиподиплоидија или речиси триплоидија | Лоша прогноза |
| Висока хипердиплоидија | Добра прогноза |
| del (9p) | Добра прогноза |
Корелација на прогнозата со цитогенетски наод на коскената срцевина кај акутна лимфобластична леукемија
| Прогноза | Цитогенетски наоди |
|---|---|
| Поволна | Хипердиплоидија > 50; t (12;21) |
| Средна | Хипердиолоидија 47 -50;
Нормално (диплоидија); del (6q); Преуредувања на 8q24 |
| Неповолна | Хиподиплоидија-близу хаплоидија; Близу тетраплоидија; del (17p); t (9;22); t (11q23) |
Се смета дека некласифицираната акутна лимфобластична леукемија има средна прогноза.[56]
Миелодиспластичен синдром
уредиМиелодиспластичниот синдром има извонредна клиничка, морфолошка и генетска хетерогеност. Цитогенетиката игра одлучувачка улога во Меѓународниот систем за прогностичко бодување на Миелодиспластичниот синдром заснован на класификацијата на Светската здравствена организација.[57][58]
- Добра прогноза: нормален кариотип, изолиран del(5q), изолиран del(20q), -Y
- Лоша прогноза: сложени абнормалности (т.е. >=3 абнормалности), -7 или del (7q)
- Средна прогноза: сите други абнормалности, вклучувајќи трисомија 8 и дел (11q)
Во споредба на метафазната цитогенетика, панелот на флуоресцентната „на лице место“ хибридизација и кариотипизацијата на низата на единечниот нуклеотиден полиморфизам за миелодиспластичниот синдром, било откриено дека секоја техника обезбедува сличен дијагностички принос. Ниту еден метод не ги откри сите дефекти, а стапките на откривање се подобрија за ~5% кога биле користени сите три методи.[59]
Стекната еднородителска дисомија, која не е забележлива со флуоресцентната „на лице место“ хибридизација или цитогенетиката, е пријавен на неколку клучни локуси во миелодиспластичниот синдром користејќи кариотипирање на низа на единечниот нуклеотиден полиморфизам, вклучително и бришење на 7/7q.[60][61]
Миелопролиферативни неоплазмени/миелопролиферативни нарушувања
уредиФиладелфиско хромозомско-негативни миелопролиферативни неоплазми вклучувајќи полицитемија вера, суштинска тромбоцитемија и примарна миелофиброза покажуваат инхерентно тежнеење за преобразба во леукемија (испуштачка фаза на миелопролиферативни неоплазми), која е придружена со стекнување на дополнителни геномски лезии. Во студијата на 159 случаи,[62] анализата на низата на единечниот нуклеотиден полиморфизам била во можност да ги долови практично сите цитогенетски абнормалности и да открие дополнителни лезии со потенцијално важни клинички последици.[се бара извор]
- Бројот на геномски промени бил повеќе од 2 до 3 пати поголем во фазата на експлозија како во хроничната фаза на болеста.
- Бришењето на 17p (TP53) било значително поврзано со претходна изложеност на хидроксиуреа, како и со сложен кариотип во примероците со испуштачка криза на миелопролиферативните неоплазми. И бришењето и неутралната загуба на хетерозиготност со копија на 17p, биле поврзани со сложен кариотип, лош прогностички маркер кај миелоидните малигни заболувања. Копирањето на неутрална загуба на хетерозиготност (стекната еднородителска дисомија) лесно може да се открие со кариотипот на низата на единечниот нуклеотиден полиморфизам, но не и од цитогенетиката, флуоресцентната „на лице место“ хибридизација или низата на споредбената геномска хибридизација.
- Пациентите во фаза на експлозија со губење на хромозомски материјал на 7q покажаа слабо преживување. Загубата на 7q е познато дека е предвидливо за брза прогресија и слаб одговор во терапијата со акутна миелоидна леукемија. Пациентите со испуштачката фаза на миелопролиферативни неоплазми со цитогенетски неоткривачки 7q копија неутрална загуба на хетерозиготност имале споредливи стапки на преживување со оние со 7/7q во нивните леукемиски клетки.
- Копирање на неутрална загуба на хетерозиготност на 9p со хомозиготна мутација на JAK2 исто така била поврзана со инфериорен исход во испуштачката фаза на миелопролиферативни неоплазми во споредба со пациентите со хетерозиготен JAK2V617F или со див вид на JAK2. За разлика од загубата на хетерозиготност на 17p, прогностичкото влијание на загубата на хетерозиготност на 9pCNN било независно од утврдените фактори на ризик како што се 7/7q, 5q или сложен кариотип.
Рак на дебелото црево
уредиИдентификацијата на биомаркерите кај ракот на дебелото црево е особено важна за пациентите со стадиум II на болеста, каде што помалку од 20% имаат повторување на туморот. Загубата на хетерозиготност на 18q е воспоставен биомаркер поврзан со висок ризик од повторување на туморот кај рак на дебелото црево со II фаза.[63]
Овие ракови се класифицирани во специфични туморски фенотипови врз основа на молекуларни профили[63] кои можат да бидат интегрирани со резултатите од други помошни тестови, како што се тестирање за нестабилност на микросателитите, имунохистохемија и статус на мутација на генот KRAS:
- Хромозомската нестабилност која има алелна нерамнотежа на голем број хромозомски локуси, вклучувајќи 5q, 8p, 17p и 18q.
- Микросателитска нестабилност која има тежнење да има диплоидни кариотипови.
Малигни рабдоидни тумори
уредиМалигните рабдоидни тумори се ретки, високо агресивни неоплазми кои најчесто се наоѓаат кај доенчиња и мали деца. Поради нивните хетерогени хистолошки одлики, дијагнозата често може да биде тешка и може да се појават погрешни класификации. Кај овие тумори, генот INI1 (SMARCB1) на хромозомот 22q функционира како класичен туморски потиснувачки ген. Исклучувањето на INI1 може да биде случено преку бришење, мутација или стекната еднородителска дисомија.[64]
Во една неодамнешна студија,[64] кариотипизацијата на низата на единечниот нуклеотиден полиморфизам идентификувала бришења или загуба на хетерозиготност од 22q во 49/51 рабдоидни тумори. Од нив, 14 биле копирање на неутрална загуба на хетерозиготност (или стекната еднородителска дисомија), што е забележливо со кариотипирање на низата на единечниот нуклеотиден полиморфизам, но не со флуоресцентната „на лице место“ хибридизација, цитогенетиката или низата со споредбената геномска хибридизација. Засилувањето со сонда зависна од мултиплекс лигатура открило едно хомозиготно бришење на еден егзон во еден примерок што беше под резолуцијата на низата на единечниот нуклеотиден полиморфизам.
Кариотипизацијата на низата на единечниот нуклеотиден полиморфизам може да биде користена за да се разликува, на пример, медулобластом со изохромозом 17q од примарен рабдоиден тумор со загуба од 22q11.2. Кога е индицирано, тогаш може да се примени молекуларна анализа на INI1 со помош на засилување со сонда зависна од мултиплекс лигатура и директно секвенционирање. Откако ќе бидат пронајдени промените поврзани со туморот, може да биде направена анализа на микробната ДНК од пациентот и родителите за да се исклучи наследна или на ново микробна мутација или бришење на INI1, за да може да бидат направени соодветни проценки на ризикот од повторување.[64]
Увеален меланом
уредиНајважната генетска промена поврзана со лошата прогноза кај увеалниот меланом е губење на цела копија од хромозомот 3 (моносомија 3), која е во силна корелација со метастатското ширење.[65] Добивките на хромозомите 6 и 8 често се користат за да се усоврши предиктивната вредност на прегледот на моносомија 3, при што добивката од 6p укажува на подобра прогноза и добивка од 8q што укажува на полоша прогноза кај туморите со дисомија 3.[66] Во ретки случаи, туморите на моносомија 3 може да ја удвојуваат преостанатата копија од хромозомот за да бидат вратени во дисомична состојба наречена изодисомија.[67] Изодисомија 3 е прогностички еквивалентна на моносомија 3, и двете може да бидат забележани со тестови за губење на хетерозиготноста на хромозомот 3.[68]
Ограничувања
уредиЗа разлика од кариотиповите добиени од конвенционалната цитогенетика, виртуелните кариотипови се реконструирани со сметачки програми користејќи сигнали добиени од „нарушена“ ДНК. Во суштина, сметачката програма ќе ги поправи транслокациите кога ќе ги подреди сигналите во хромозомски редослед. Затоа, виртуелните кариотипови не можат да забележуваат урамнотежени транслокации и инверзии. Тие, исто така, можат да забележуваат генетски аберации само во регионите на геномот што се претставени со сонди на низата. Покрај тоа, виртуелните кариотипови создаваат „релативен“ број на копии нормализирани во однос на диплоиден геном, така што тетраплоидните геноми ќе се кондензираат во диплоиден простор освен ако не биде извршена ренормализација. Ренормализацијата бара помошна анализа засновата на клетки, како што е флуоресцентната „на лице место“ хибридизација, ако некој користи низа на споредбена геномска хибридизација. За кариотиповите добиени од низи базирани на единечен нуклеотиден полиморфизам, тетраплоидијата често може да биде заклучена од одржувањето на хетерозиготноста во регионот на очигледна загуба на бројот на копии.[22] Мозаицизам на ниско ниво или мали подклонови може да не бидат откриени со виртуелни кариотипови бидејќи присуството на нормални клетки во примерокот ќе го намали сигналот од абнормалниот клон. Точната точка на неуспех, во однос на минималниот процент на неопластични клетки, ќе зависи од одредената платформа и алгоритмите што се користени. Многу софтверски програми за анализа на бројот на копии што се користени за создавање на кариотипови засновани на низа ќе попуштат со помалку од 25-30% туморни/абнормални клетки во примерокот. Меѓутоа, во онколошките примени ова ограничување може да биде минимизирано со стратегии за збогатување на туморот и софтвер оптимизиран за употреба со онколошки примероци. Алгоритмите за анализа се развивани брзо, а некои се дизајнирани дури и да напредуваат на „нормална контаминација на клонови“,[69] па се очекува дека ова ограничување ќе продолжи да исчезнува.
Поврзано
уреди- DECIPHER, база на податоци за хромозомска нерамнотежа и фенотип кај луѓето кои користат ресурси на Ensembl
Наводи
уреди- ↑ Digital karyotyping - Wang et al., 10.1073/pnas.202610899 - Proceedings of the National Academy of Sciences
- ↑ „The array CGH and its clinical applications“. Drug Discov Today. 13 (17–18): 760–70. 2008. doi:10.1016/j.drudis.2008.06.007. PMID 18617013.
- ↑ White M.J.D. 1973. The chromosomes. 6th ed, Chapman & Hall, London. p28
- ↑ Stebbins G.L. 1950. Variation and evolution in plants. Chapter XII: The Karyotype. Columbia University Press N.Y.
- ↑ Shaffer, LG; Bejjani, BA. (2006). „Medical applications of array CGH and the transformation of clinical cytogenetics“. Cytogenet. Genome Res. 115 (3–4): 303–9. doi:10.1159/000095928. PMID 17124414.
- ↑ Edelmann, L; Hirschhorn, K. (јануари 2009). „Clinical utility of array CGH for the detection of chromosomal imbalances associated with mental retardation and multiple congenital anomalies“. Annals of the New York Academy of Sciences. 1151 (1): 157–66. Bibcode:2009NYASA1151..157E. doi:10.1111/j.1749-6632.2008.03610.x. PMID 19154522.
- ↑ Dutt, A; Beroukhim, R. (јануари 2007). „Single nucleotide polymorphism array analysis of cancer“. Current Opinion in Oncology. 19 (1): 43–9. doi:10.1097/CCO.0b013e328011a8c1. PMID 17133111.
- ↑ 8,0 8,1 8,2 Hagenkord JM; Parwani AV; Lyons-Weiler MA; Alvarez K; Amato R; Gatalica Z; Gonzalez-Berjon JM; Peterson L; Dhir R (ноември 2008). „Virtual karyotyping with SNP microarrays reduces uncertainty in the diagnosis of renal epithelial tumors“. Diagn Pathol. 3 (1): 44. doi:10.1186/1746-1596-3-44. PMC 2588560. PMID 18990225.
- ↑ Beaudet, AL; Belmont, JW. (2008). „Array-based DNA diagnostics: let the revolution begin“. Annu Rev Med. 59 (1): 113–29. doi:10.1146/annurev.med.59.012907.101800. PMID 17961075.
- ↑ 10,0 10,1 Monzon FA; Hagenkord JM; Lyons-Weiler MA; Balani JP; Parwani AV; Sciulli CM; Li J; Chandran UR; Bastacky SI (мај 2008). „Whole genome SNP arrays as a potential diagnostic tool for the detection of characteristic chromosomal aberrations in renal epithelial tumors“. Mod Pathol. 21 (5): 599–608. doi:10.1038/modpathol.2008.20. PMID 18246049.
- ↑ Leary RJ; Lin JC; Cummins J; Boca S; Wood LD; Parsons DW; Jones S; Sjöblom T; Park BH (2008). „Integrated analysis of homozygous deletions, focal amplifications, and sequence alterations in breast and colorectal cancers“. Proc Natl Acad Sci U S A. 105 (42): 16224–9. Bibcode:2008PNAS..10516224L. doi:10.1073/pnas.0808041105. PMC 2571022. PMID 18852474.
- ↑ 12,0 12,1 Lehmann S; Ogawa S; Raynaud SD; Sanada M; Nannya Y; Ticchioni M; Bastard C; Kawamata N; Koeffler HP (март 2008). „Molecular allelokaryotyping of early-stage, untreated chronic lymphocytic leukemia“. Cancer. 112 (6): 1296–305. doi:10.1002/cncr.23270. PMID 18246537.
- ↑ Vermeesch JR; Fiegler H; de Leeuw N; Szuhai K; Schoumans J; Ciccone R; Speleman F; Rauch A; Clayton-Smith J (November 2007). „Guidelines for molecular karyotyping in constitutional genetic diagnosis“. Eur J Hum Genet. 15 (11): 1105–14. doi:10.1038/sj.ejhg.5201896. PMID 17637806.
- ↑ Kulharya, AS; Flannery, DB; Norris, K; Lovell, C; Levy, B; Velagaleti, GV. (септември 2008). „Fine mapping of breakpoints in two unrelated patients with rare overlapping interstitial deletions of 9q with mild dysmorphic features“. American Journal of Medical Genetics. 146A (17): 2234–41. doi:10.1002/ajmg.a.32397. PMID 18666229.
- ↑ Nowakowska B; Stankiewicz P; Obersztyn E; Ou Z; Li J; Chinault AC; Smyk M; Borg K; Mazurczak T (септември 2008). „Application of metaphase HR-CGH and targeted Chromosomal Microarray Analyses to genomic characterization of 116 patients with mental retardation and dysmorphic features“. American Journal of Medical Genetics. 146A (18): 2361–9. doi:10.1002/ajmg.a.32475. PMID 18698622.
- ↑ Probst FJ; Roeder ER; Enciso VB; Ou Z; Cooper ML; Eng P; Li J; Gu Y; Stratton RF (јуни 2007). „Chromosomal microarray analysis (CMA) detects a large X chromosome deletion including FMR1, FMR2, and IDS in a female patient with mental retardation“. American Journal of Medical Genetics. 143A (12): 1358–65. doi:10.1002/ajmg.a.31781. PMID 17506108.
- ↑ Hildebrandt, F; и др. (јануари 2009). „A systematic approach to mapping recessive disease genes in individuals from outbred populations“. PLOS Genet. 5 (1): e1000353. doi:10.1371/journal.pgen.1000353. PMC 2621355. PMID 19165332.
- ↑ McQuillan R; Leutenegger AL; Abdel-Rahman R; Franklin CS; Pericic M; Barac-Lauc L; Smolej-Narancic N; Janicijevic B; Polasek O (2008). „Runs of homozygosity in European populations“. Am J Hum Genet. 83 (3): 359–72. doi:10.1016/j.ajhg.2008.08.007. PMC 2556426. PMID 18760389.
- ↑ Gondek, LP; Tiu, R; O'Keefe, CL; Sekeres, MA; Theil, KS; Maciejewski, JP. (февруари 2008). „Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS-derived AML“. Blood. 111 (3): 1534–42. doi:10.1182/blood-2007-05-092304. PMC 2214746. PMID 17954704.
- ↑ Beroukhim R; Lin M; Park Y; Hao K; Zhao X; Garraway LA; Fox EA; Hochberg EP; Mellinghoff IK (мај 2006). „Inferring loss-of-heterozygosity from unpaired tumors using high-density oligonucleotide SNP arrays“. PLOS Comput. Biol. 2 (5): e41. Bibcode:2006PLSCB...2...41B. doi:10.1371/journal.pcbi.0020041. PMC 1458964. PMID 16699594.
- ↑ Ishikawa S; Komura D; Tsuji S; Nishimura K; Yamamoto S; Panda B; Huang J; Fukayama M; Jones KW (август 2005). „Allelic dosage analysis with genotyping microarrays“. Biochem Biophys Res Commun. 333 (4): 1309–14. doi:10.1016/j.bbrc.2005.06.040. PMID 15982637.
- ↑ 22,0 22,1 Lo, KC; Bailey, D; Burkhardt, T; Gardina, P; Turpaz, Y; Cowell, JK. (март 2008). „Comprehensive analysis of loss of heterozygosity events in glioblastoma using the 100K SNP mapping arrays and comparison with copy number abnormalities defined by BAC array comparative genomic hybridization“. Genes Chromosomes Cancer. 47 (3): 221–37. doi:10.1002/gcc.20524. PMID 18050302.
- ↑ Mao, X; Young, BD; Lu, YJ. (јуни 2007). „The application of single nucleotide polymorphism microarrays in cancer research“. Curr Genomics. 8 (4): 219–28. doi:10.2174/138920207781386924. PMC 2430687. PMID 18645599.
- ↑ „Overall genomic pattern is a predictor of outcome in neuroblastoma“. J. Clin. Oncol. 27 (7): 1026–33. март 2009. doi:10.1200/JCO.2008.16.0630. PMID 19171713.
- ↑ Michels, E; Vandesompele, J; Hoebeeck, J; Menten, B; De Preter, K; Laureys, G; Van Roy, N; Speleman, F. (2006). „Genome wide measurement of DNA copy number changes in neuroblastoma: dissecting amplicons and mapping losses, gains and breakpoints“. Cytogenet. Genome Res. 115 (3–4): 273–282. doi:10.1159/000095924. PMID 17124410.
- ↑ Messahel B; Williams R; Ridolfi A; A'hern R; Warren W; Tinworth L; Hobson R; Al-Saadi R; Whyman G (март 2009). „Children's Cancer and Leukaemia Group (CCLG). Allele loss at 16q defines poorer prognosis Wilms tumour irrespective of treatment approach in the UKW1-3 clinical trials: a Children's Cancer and Leukaemia Group (CCLG) Study“. Eur J Cancer. 45 (5): 819–26. doi:10.1016/j.ejca.2009.01.005. PMID 19231157.
- ↑ Grundy PE; Breslow NE; Li S; Perlman E; Beckwith JB; Ritchey ML; Shamberger RC; Haase GM; D'Angio GJ (октомври 2005). „National Wilms Tumor Study Group. Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms tumor: a report from the National Wilms Tumor Study Group“. J Clin Oncol. 23 (29): 7312–21. doi:10.1200/JCO.2005.01.2799. PMID 16129848.
- ↑ van den Berg, E; Störkel, S (2003). „Kidney: Clear cell renal cell carcinoma“. Atlas Genet Cytogenet Oncol Haematol. 7 (3): 424–431. Архивирано од изворникот на 2011-04-25. Посетено на 14 December 2010.
- ↑ Lyons-Weiler, MA; Hagenkord, JM; Sciulli, CM; Dhir, R; Monzon, FA. (2008). „Optimization of the Affymetrix GeneChip Mapping 10K 2.0 Assay for Routine Clinical Use on Formalin Fixed Paraffin Embedded Tissues“. Diag Mol Path. 17 (1): 3–13. doi:10.1097/PDM.0b013e31815aca30. PMID 18303412.
- ↑ Klatte T; Pantuck AJ; Said JW; Seligson DB; Rao NP; LaRochelle JC; Shuch B; Zisman A; Kabbinavar FF (2009). „Cytogenetic and molecular tumor profiling for type 1 and type 2 papillary renal cell carcinoma“. Clinical Cancer Research. 15 (4): 1162–9. doi:10.1158/1078-0432.CCR-08-1229. PMID 19228721.
- ↑ Brunelli M; Eccher A; Gobbo S; Ficarra V; Novara G; Cossu-Rocca P; Bonetti F; Menestrina F; Cheng L (2008). „Loss of chromosome 9p is an independent prognostic factor in patients with clear cell renal cell carcinoma“. Modern Pathology. 21 (1): 1–6. doi:10.1038/modpathol.3800967. PMID 17906617.
- ↑ Klatte T; Rao PN; de Martino M; LaRochelle J; Shuch B; Zomorodian N; Said J; Kabbinavar FF; Belldegrun AS (2009). „Cytogenetic profile predicts prognosis of patients with clear cell renal cell carcinoma“. Journal of Clinical Oncology. 27 (5): 746–53. doi:10.1200/JCO.2007.15.8345. PMID 19124809.
- ↑ Szponar, A; Zubakov, D; Pawlak, J; Jauch, A; Kovacs, G (2009). „Three genetic developmental stages of papillary renal cell tumors: duplication of chromosome 1q marks fatal progression“. International Journal of Cancer. 124 (9): 2071–6. doi:10.1002/ijc.24180. PMID 19123481.
- ↑ Schwaenen C; Nessling M; Wessendorf S; Salvi T; Wrobel G; Radlwimmer B; Kestler HA; Haslinger C; Stilgenbauer S (2004). „Automated array-based genomic profiling in chronic lymphocytic leukemia: development of a clinical tool and discovery of recurrent genomic alterations“. Proc Natl Acad Sci U S A. 101 (4): 1039–44. Bibcode:2004PNAS..101.1039S. doi:10.1073/pnas.0304717101. PMC 327147. PMID 14730057.
- ↑ Pfeifer D; Pantic M; Skatulla I; Rawluk J; Kreutz C; Martens UM; Fisch P; Timmer J; Veelken H (февруари 2007). „Genome-wide analysis of DNA copy number changes and LOH in CLL using high-density SNP arrays“. Blood. 109 (3): 1202–10. doi:10.1182/blood-2006-07-034256. PMID 17053054.
- ↑ Gunn SR; Mohammed MS; Gorre ME; Cotter PD; Kim J; Bahler DW; Preobrazhensky SN; Higgins RA; Bolla AR (септември 2008). „Whole-genome scanning by array comparative genomic hybridization as a clinical tool for risk assessment in chronic lymphocytic leukemia“. The Journal of Molecular Diagnostics. 10 (5): 442–451. doi:10.2353/jmoldx.2008.080033. PMC 2518739. PMID 18687794.
- ↑ Sargent R; Jones D; Abruzzo LV; Yao H; Bonderover J; Cisneros M; Wierda WG; Keating MJ; Luthra R (јануари 2009). „Customized oligonucleotide array-based comparative genomic hybridization as a clinical assay for genomic profiling of chronic lymphocytic leukemia“. J Mol Diagn. 11 (1): 25–34. doi:10.2353/jmoldx.2009.080037. PMC 2607562. PMID 19074592.
- ↑ 2009 мај ;23(5):829-33
- ↑ Hagenkord, JM; Monzon, FA; Kash, SF; Lilleberg, S; Xie, Q; Kant, JA (2010). „Array-based karyotyping for prognostic assessment in chronic lymphocytic leukemia: performance comparison of affymetrix 10K2.0, 250K Nsp, and SNP6.0 arrays“. J Mol Diagn. 12 (2): 184–96. doi:10.2353/jmoldx.2010.090118. PMC 2871725. PMID 20075210.
- ↑ „Genomic aberrations and survival in chronic lymphocytic leukemia“. NEJM. 343 (26): 1910–6. 2000. doi:10.1056/NEJM200012283432602. PMID 11136261.
- ↑ Hervé Avet-Loiseau; Cheng Li; Florence Magrangeas; Wilfried Gouraud; Catherine Charbonnel; Jean-Luc Harousseau; Michel Attal; Gerald Marit; Claire Mathiot (септември 2009). „Prognostic significance of copy-number alterations in multiple myeloma“. Journal of Clinical Oncology. 27 (27): 4585–90. doi:10.1200/JCO.2008.20.6136. PMC 2754906. PMID 19687334.
- ↑ Pfister S; Remke M; Benner A; Mendrzyk F; Toedt G; Felsberg J; Wittmann A; Devens F; Gerber NU (април 2009). „Outcome Prediction in Pediatric Medulloblastoma based on DNA Copy Number Aberrations of Chromosomes 6q and 17q and the MYC and MYCN Loci“. J Clin Oncol. 27 (10): 1627–1636. doi:10.1200/JCO.2008.17.9432. PMID 19255330.
- ↑ 43,0 43,1 „Allelic losses at 1p36 and 19q13 in gliomas: correlation with histologic classification, definition of a 150-kb minimal deleted region on 1p36, and evaluation of CAMTA1 as a candidate tumor suppressor gene“. Clin. Cancer Res. 11 (3): 1119–28. 1 февруари 2005. doi:10.1158/1078-0432.1119.11.3. PMID 15709179.
- ↑ „[Molecular biology of oligodendroglial tumors]“. Neuro-Chirurgie (француски). 51 (3–4 Pt 2): 260–8. 2005. doi:10.1016/s0028-3770(05)83487-3. PMID 16292170.
- ↑ „Clinical use of genotype to predict chemosensitivity in oligodendroglial tumors“. Neurology. 66 (11): 1661–7. 2006. doi:10.1212/01.wnl.0000218270.12495.9a. PMID 16769937.
- ↑ „A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma“. Cancer Res. 66 (20): 9852–61. октомври 2006. doi:10.1158/0008-5472.CAN-06-1796. PMID 17047046.
- ↑ „p53 mutations in nonastrocytic human brain tumors“. Cancer Res. 51 (22): 6202–5. 15 ноември 1991. PMID 1933879.
- ↑ „Anaplastic oligodendrogliomas with 1p19q codeletion have a proneural gene expression profile“. Mol. Cancer. 7 (1): 41. 2008. doi:10.1186/1476-4598-7-41. PMC 2415112. PMID 18492260.
- ↑ Dong Yin; Seishi Ogawa; Norihiko Kawamata; Patrizia Tunici; Gaetano Finocchiaro; Marica Eoli; Christian Ruckert; Thien Huynh; Gentao Liu (мај 2009). „High-Resolution Genomic Copy Number Profiling of Glioblastoma Multiforme by Single Nucleotide Polymorphism DNA Microarray“. Mol Cancer Res. 7 (5): 665–77. doi:10.1158/1541-7786.MCR-08-0270. PMID 19435819.
- ↑ Cancer Cytogenetics, 3rd Ed, Chapter 19, Tumors of the Nervous System, Wiley Blackwell 2009.
- ↑ Tumors of the Central Nervous System. Vol 7. Washington DC: American Registry of Pathology; 2007
- ↑ „Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial“. Blood. 109 (8): 3189–97. 2007. doi:10.1182/blood-2006-10-051912. PMID 17170120.
- ↑ Kawamata N; Ogawa S; Zimmermann M; Kato M; Sanada M; Hemminki K; Yamatomo G; Nannya Y; Koehler R (јануари 2008). „Molecular allelokaryotyping of pediatric acute lymphoblastic leukemias by high-resolution single nucleotide polymorphism oligonucleotide genomic microarray“. Blood. 111 (2): 776–84. doi:10.1182/blood-2007-05-088310. PMC 2200831. PMID 17890455.
- ↑ Bungaro S; Dell'Orto MC; Zangrando A; Basso D; Gorletta T; Lo Nigro L; Leszl A; Young BD; Basso G (јануари 2009). „Integration of genomic and gene expression data of childhood ALL without known aberrations identifies subgroups with specific genetic hallmarks“. Genes Chromosomes Cancer. 48 (1): 22–38. doi:10.1002/gcc.20616. PMID 18803328.
- ↑ Sulong S; Moorman AV; Irving JA; Strefford JC; Konn ZJ; Case MC; Minto L; Barber KE; Parker H (јануари 2009). „A comprehensive analysis of the CDKN2A gene in childhood acute lymphoblastic leukemia reveals genomic deletion, copy number neutral loss of heterozygosity, and association with specific cytogenetic subgroups“. Blood. 113 (1): 100–7. doi:10.1182/blood-2008-07-166801. PMID 18838613.
- ↑ „A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study“. Lancet Oncol. 10 (2): 125–34. јануари 2009. doi:10.1016/S1470-2045(08)70339-5. PMC 2707020. PMID 19138562.
- ↑ Hasse, D (2008). „Cytogenetic features in myelodysplastic syndromes“. Ann Hematol. 87 (7): 515–526. doi:10.1007/s00277-008-0483-y. PMC 2413090. PMID 18414863.
- ↑ WHO Classification of Tumours of Haematopoeitic and Lymphoid Tissues, Edited by Swerdlow SH, et al. IARC Press, 2008, Lyon.
- ↑ Makishima H; Rataul M; Gondek LP; Huh J; Cook JR; Theil KS; Sekeres MA; Kuczkowski E; O'Keefe C (2010). „FISH and SNP-A karyotyping in myelodysplastic syndromes: Improving cytogenetic detection of del(5q), monosomy 7, del(7q), trisomy 8, and del(20q)“. Leuk Res. 34 (4): 447–453. doi:10.1016/j.leukres.2009.08.023. PMC 2826525. PMID 19758696.
- ↑ Sanada, et al. "Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms." Nature 13 август 2009; 460, 904–909.
- ↑ Gondek, LP; Tiu, R; O'Keefe, CL; Sekeres, MA; Theil, KS; MacIejewski, JP (2008). „Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS-derived AML“. Blood. 111 (3): 1534–42. doi:10.1182/blood-2007-05-092304. PMC 2214746. PMID 17954704.
- ↑ Thoennissen NH; Krug UO; Lee DH; Kawamata N; Iwanski GB; Lasho T; Weiss T; Nowak D; Koren-Michowitz M (април 2010). „Prevalence and prognostic impact of allelic imbalances associated with leukemic transformation of Philadelphia chromosome-negative myeloproliferative neoplasms“. Blood. 115 (14): 2882–2890. doi:10.1182/blood-2009-07-235119. PMC 2854432. PMID 20068225.
- ↑ 63,0 63,1 Lenz HJ, "Established Biomarkers for Colorectal Carcinoma", American Society of Clinical Oncology Educational Book, 2009, p215-219.
- ↑ 64,0 64,1 64,2 Jackson EM; Sievert AJ; Gai X; Hakonarson H; Judkins AR; Tooke L; Perin JC; Xie H; Shaikh TH (2009). „Genomic analysis using high density single nucleotide polymorphism-based oligonucleotide arrays and multiplex ligation-dependent probe amplification provides comprehensive analysis of INI1/SMARCB1 in Malignant Rhabdoid Tumors“. Clin Cancer Res. 15 (6): 1923–1930. doi:10.1158/1078-0432.CCR-08-2091. PMC 2668138. PMID 19276269.
- ↑ „Prognostic implications of monosomy 3 in uveal melanoma“. Lancet. 347 (9010): 1222–1225. 1996. doi:10.1016/S0140-6736(96)90736-9. PMID 8622452.
- ↑ „Multiplex Ligation-Dependent Probe Amplification of Uveal Melanoma: Correlation with Metastatic Death“ (PDF). Invest Ophthalmol Vis Sci. 50 (7): 3048–55. 2009. doi:10.1167/iovs.08-3165. PMID 19182252.
|hdl-access=бара|hdl=(help) - ↑ „Acquired homozygosity (isodisomy) of chromosome 3 in uveal melanoma“. Cancer Genet Cytogenet. 102 (1): 40–45. 1998. doi:10.1016/S0165-4608(97)00290-2. PMID 9530338.
- ↑ „Loss of heterozygosity of chromosome 3 detected with single nucleotide polymorphisms is superior to monosomy 3 for predicting metastasis in uveal melanoma“. Clin Cancer Res. 13 (10): 2923–2937. 2007. doi:10.1158/1078-0432.CCR-06-2383. PMID 17504992.
- ↑ Yamamoto G; Nannya Y; Kato M; Sanada M; Levine RL; Kawamata N; Hangaishi A; Kurokawa M; Chiba S (јули 2007). „Highly sensitive method for genomewide detection of allelic composition in nonpaired, primary tumor specimens by use of affymetrix single-nucleotide-polymorphism genotyping microarrays“. Am J Hum Genet. 81 (1): 114–26. doi:10.1086/518809. PMC 1950910. PMID 17564968.